
Translate this page into:
An N-terminal peptide of Tar DNA binding Protein 43 lacking nuclear localization signal translocates to the nucleus of GC-1 spermatogonial cells


*Corresponding author: Pradeep G. Kumar, Scientist ‘G’, Rajiv Gandhi Centre for Biotechnology, Thiruvananthapuram, Kerala, India. kumarp@rgcb.res.in
-
Received: ,
Accepted: ,
How to cite this article: Varghese DS, Vysakh G, Kumar PG. An N-terminal peptide of Tar DNA Binding Protein 43 lacking nuclear localization signal translocates to the nucleus of GC-1 spermatogonial cells. J Reprod Healthc Med 2023;4:3.
Abstract
Objectives:
TAR DNA-binding protein of 43 kDa (TDP-43) is an RNA/DNA binding protein expressed in the brain and the testis. Mutations in TDP-43 lead to mislocalization and cytoplasmic aggregation of this protein causing neurodegenerative diseases such as amyotrophic lateral sclerosis and frontotemporal dementia. TDP-43 has also been implicated in maintaining spermatogenesis. While homodimerization of TDP-43 is critical for its physiological functions, higher-order aggregation of this protein impairs its functions. This study was aimed to map the critical amino acids of the N-terminus of this protein in mediating its homodimerization.
Materials and Methods:
We generated deletion constructs of Tdp-43 containing NRRM1 domain alone (TDP-43∆3-183) and N-terminal peptide of TDP-43 which lacks the nuclear localization signal (NLS) (TDP-43∆1-50) with fluorescent reporters having non-overlapping emission properties. These constructs were co-transfected into a mouse spermatogonial cell line to examine their dimerization and nuclear translocation capabilities in vitro.
Results:
We found that TDP-43∆3-183 alone was not capable of homodimerization. On the other hand, TDP-43∆1-50 when co-transfected into GC1-spg cells along with full length TDP-43 translocated to the nucleus oligomerized with the latter and translocated to the nucleus, indicating the importance of amino acids 1-50 of TDP-43 in dimerization.
Conclusion:
The N-terminal segment of TDP-43 spanning amino acids 1-50 is responsible for dimerization, while that spanning amino acids 51-183 directs it to the nucleus.The physiological and pathological implications of this finding need to be examined.
Keywords
TAR DNA binding protein
Dimerization
Testis
Spermatogenesis
Mislocalization
Protein aggregation

INTRODUCTION
TAR DNA binding protein-43 (TDP-43) is a transcriptional repressor that binds to the transactive response element DNA sequence of the viral genome and is indispensable for the regulation of the viral gene associated with HIV-1 transcription.[1] It belongs to the heterogeneous ribonucleoprotein (hnRNP) family and binds to RNA/DNA with significant sequence-specificity using one or more highly conserved RNA recognition motifs (RRM1 and RRM2).[2] The presence of two octameric and hexameric sequences of RRM1, namely, RNP1 and RNP2, also aids in alternative splicing. The N-terminal domain (NTD) spans amino acids (AA) 4–77 with bipartite nuclear localization signals (NLS) and three caspase-3 cleavage sites.[3] It also has nuclear export signal (NES) and a glycine-rich C-terminal domain (CTD)[4] that facilitates exon skipping[5] and protein-protein interaction between TDP-43 and other hnRNP members.[6]
The N-terminal residues play a crucial role in the dimerization of TDP-43 which, in turn, is critical for its physiological functions such as RNA splicing.[7] Any mutation within the NLS signal or that affects the caspase cleavage at Asp 89 leads to the accumulation of mislocalized TDP-43 in the cytoplasm.[8] The NES at AA 239–250 of TDP-43 lies within the folded globular RRM domain.[9] Mutations in the CTD are also associated with the pathological behavior of mislocalized TDP-43.[10]
One of the unique functions of TDP-43 is the self-regulation of its mRNA or maintaining the stability of its mRNAs and several other mRNAs by directly interacting with their transcripts, possibly through their 3’UTR regions.[11] Depletion of TDP-43 in the nucleus can affect its splicing function.[12] However, TDP-43 has a role in regulating the splicing patterns of mRNA of many significant genes, such as Cystic fibrosis transmembrane conductance regulator (CFTR), TARDBP, FUS, SNCA (α-synuclein), HTT (Huntingtin), and Amyloid precursor protein.[7]
The mislocalized TDP-43 in the central nervous system is associated with many neurodegenerative diseases such as Amyotrophic Lateral Sclerosis, Frontotemporal Lobar Degeneration, Alzheimer’s disease, and Limbic Predominant Age-related TDP-43 Encephalopathy. Even though the medical ailments of these diseases are different, they show a comparable pathological condition.[13] In addition, TDP-43 undergoes major post-translational modifications such as cleavage, hyperphosphorylation, and ubiquitination under pathological conditions.[11] These modifications cause the protein to accumulate and aggregate as inclusion bodies in the cytoplasm. All the neurodegenerative diseases mentioned above manifest sequential TDP-43 aggregation in the different regions of the brain during the disease progression [Table 1].[14-17]
| Condition | TDP-43 deposition in the brain | |||||
|---|---|---|---|---|---|---|
| Stage 1 | Stage 2 | Stage 3 | Stage 4 | Stage 5 | Stage 6 | |
| AD | Amygdala | Entorhinal cortex and subiculum | Dentate gyrus of the hippocampus and occipitotemporal cortex | Insular cortex, ventral striatum, basal forebrain and inferior temporal cortex | Substantia nigra, inferior olive and midbrain tectum | Basal ganglia and middle frontal cortex |
| FTLD | Prefrontal neocortex and amygdala | Thalamus motor cortex | Neocortical areas, and spinal cord | Occipital neocortex region and the visual processing center of the brain | ||
| ALS | Projection neurons of the agranular motor cortex, brainstem motor nuclei of cranial nerves V, VII, and X–XII and spinal cord | Middle frontal gyrus, reticular formation, precerebellar nuclei of the brainstem, and the red nucleus | Prefrontal (gyrus rectus and orbital gyri), postcentral neocortex striatum, and basal ganglia | Anteromedial areas of the temporal lobe, entorhinal cortex, hippocampal and dentate fascia | ||
| LATE-NC | Amygdala | Hippocampus | Middle frontal gyrus | |||
TDP-43 is expressed predominantly in the testes as well. The expression of TDP-43 was detected in the nuclei of both germ cells and Sertoli cells of the seminiferous epithelium. In the germ cells, its expression was detectable in type B spermatogonia.[18] Interestingly, in the testis, it acts as a transcriptional repressor of the Acrv1 gene, which translates the acrosomal protein, SP-10, during spermatogenesis.[19] Aberrant expression of TDP-43 in the testis has been associated with infertility in men.[20] In line with this, a loss of function of TDP-43 in male mouse models resulted in a meiotic arrest in the process of spermatogenesis.[21]
The homodimerization and nuclear localization of TDP-43 are assigned to AA 4-77. However, the minimal essential domain of TDP-43 that exhibits the homodimerization capability is not known. We provide evidence to show that a deletion construct of TDP-43 containing the AA 1–50 was incompetent to be targeted to the nucleus on its own, yet could relocate to the nucleus by associating with full length TDP-43. Thus, we provide experimental evidence to state that the homodimerization domain of TDP-43 lies within AA 1–50, while the nuclear localization domain appears to be located posterior to this region.
MATERIALS AND METHODS
Reagents
Acrylamide, Agarose, Ammonium persulphate, Ampicillin, β-mercaptoethanol, Bovine Serum Albumin (BSA), Boric acid, 3-([3-Chloroamidopropyl] dimethylammonio)-1-propanesulfonate, Chloroform, 3,3`Diaminobenzidinetetrachloride hydrate (DAB) Dispase, Dithiothreitol, Ethidium bromide, Ethylenediamenetetraacetic acid (EDTA), Ethyleneglycoltetracetic acid, Gelatin, Glycerol, Glycine, HEPES, Heat inactivated foetal Bovine Serum (HI-FBS), Hoechst 33342, Igepal (NP-40), Imidazole, Isopropanol, Isopropyl β-D-1-thiogalactopyranoside (IPTG), Kanamycin, Magnesium chloride (MgCl2), N, N`-Methylene bis acrylamide, phenylmethylsulfonyl fluoride, Poly-L-lysine, Primers, Propidium Iodide (PI), Protease inhibitor cocktail, Sodium bicarbonate, Sodium chloride (NaCl), Sodium dodecyl sulphate (SDS), Sodium hydroxide, Sodium orthovanadate, TRI reagent, Tris Hydrochloric acid (HCl), Trizma base, Triton ×−100, Trypsin EDTA, Tween 20, and Urea and Rabbit polyclonal anti-TARDBP-1 antibody (Cat.No.AV38941) were from Sigma-Aldrich, MO, USA. DNA ladders, ×10 polymerase chain reaction (PCR) buffer, HindIII, SacII, SalI, T4 DNA ligase, and Taq DNA polymerase were from New England Biolabs, MA, USA. Acetic acid, Bromophenol Blue, Calcium chloride, Coomassie Brilliant Blue-R-250, n-Butanol, Nickel chloride, Paraformaldehyde, potassium dihydrogen phosphate (KH2PO4), potassium chloride (KCl), Sodium acetate, and Xylene cyanol were from SR Laboratories, Mumbai, India. Agar, Beef extract, Sodium deoxycholate, Tryptone, and Yeast extract were from HiMedia, Mumbai, India. Antibiotic-antimycotic, Dulbecco’s Modified Eagle Medium, FBS and GC-1 SPG cells (ATCC® CRL 2053™) were from ATCC, VA, USA. Deoxynucleotide triphosphates and Polyvinylidine fluoride (PVDF) membrane, GelBond PAG film, Illustra plasmidPrep Mini Spin Kit, Illustra GFX PCR DNA, and GelBand Purification Kit were from GE Healthcare, NA, UK. Disodium hydrogen phosphate (Na2HPO4) was from Qualigens fine chemicals, Mumbai, India. DPX mountant, Paraffin, and Xylene were from Merck Millipore, MA, USA. Molecular biology grade ethanol was from Brampton, Ontario, Canada. HCl, Hydrogen peroxide, and Sulfuric acid were sourced from SD fine chemicals, Mumbai, India. Non-fat milk powder has been bought from Sagar, Gujarat, India. Lipofetamine® 2000, Opti-MEM® I Reduced Serum Medium, pET 100/D vector and pET 102/D vector and Alexa fluor® 488 conjugate (Cat. No. A-11008) were from Invitrogen, CA, USA. Mammalian expression vectors EGFP-C1, pEGFP-N1, and pZsYellow1-C were purchased from Clontech, CA, USA. Precision Markers were bought from Bio-RAD Laboratories, CA, USA. Big Dye Terminator v3.1 Cycle Sequencing Kit was from AB Applied Biosystems, CA, USA. ChampionTM pET Directional TOPO® expression kit (Catalog Nos K100-01 and K102-01) and SuperScript® VILO™ cDNA Synthesis Kit were from Invitrogen, CA, USA. Ready-To-Go T- prime first strand synthesis kit was from Amersham Biosciences NJ, USA. Goat polyclonal anti β-ACTIN antibody (Cat.No. sc-1616), Goat anti-rabbit IgG-HRP antibody (Cat.No. sc-2030), Rabbit anti-goat IgG-HRP antibody (Cat.No. sc-2768), and Donkey anti-mouse IgG-HRP antibody (Cat.No. sc-2318) were purchased from Santa Cruz Biotechnology Inc. CA, USA. Rabbit polyclonal anti-TDP-43 (G400) antibody (Cat. No. 3448S) was purchased from Cell Signaling Technology, MA, USA. Goat anti-rabbit IgG H&L (FITC) (Cat.No. ab6717) and Goat anti-rabbit IgG H&L (TRITC) (Cat No. ab 6718) were from Abcam, Cambridge.
RNA isolation, cDNA synthesis and Reverse transcriptase polymerase chain reaction
RNA was isolated using the Trizol method and the cDNA was synthesized using Ready- To-Go T-Primed First-Strand Kit or SuperScript® VILO™ cDNA Synthesis Kit as per the manufacturer’s instructions. The cDNA thus produced was used for PCR amplification in a Veriti® thermal cycler (Applied Biosystems, CA, USA). Primer Blast (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) was used to design primers using Tdp-43 sequences in murine brain available in the NCBI database (http://www.ncbi.nlm.nih. gov/BLAST/). The primer sequences used are tabulated in [Table S1]. The PCR products were electrophoresed on agarose gels and the gel image was captured using the VersaDoc imaging system (BioRad, USA). The desired bands were cut and eluted from agarose gels using Illustra GFX PCR DNA and GelBAnd Purification kit following the manufacturer’s instructions.
TA cloning and automated sequencing
Tardbp CDS was cloned into the TOPO TA vector following the TOPO TA cloning kit protocol (Invitrogen, CA, USA). The automated sequencing reaction was done using the Big Dye Terminator v3.1 Cycle sequencing kit as per the manufacturer’s protocol. The dye terminated products were precipitated and were sequenced in an automated DNA sequencing machine (Thermo fisher scientific Waltham, Massachusetts, United States). The sequences were then analyzed using NCBI nucleotide BLAST.
Directional cloning, expression and purification of TDP- 43∆1-50 and TDP-43∆3-183 into pET 102/D- TOPO® or pET 100/D-TOPO® expression vector
Transcript variant 1 of Tardbp in TA plasmid was used as the template for directional cloning. TDP-43∆1-50 was cloned into the pET 102/D-TOPO vector, whereas TDP-43∆3-183 was cloned into the pET 100/D-TOPO® expression vector (Invitrogen, CA, USA) following the manufacturer’s protocol. The expression of recombinant protein in BL21 (DE3) cells was induced using 1M IPTG for both the vectors. The soluble proteins in the uninduced and induced lysates were extracted using lysis buffer (50 mM potassium phosphate, pH 7.8, 400 mM NaCl, 100 mM KCl, 10% glycerol, 0.5% Triton ×−100, 10 mM imidazole). Insoluble protein and the protein secluded within inclusion were extracted using Laemmli buffer (0.5 M Tris-HCl, pH 6.8, 20% glycerol, 10% β-mercaptoethanol, 4% SDS and 0.005% bromophenol blue). The crude protein lysates were purified using His Trap FF crude column and the elutes were collected as 1 mL fractions by passing 5 mL each of 100–500 mM imidazole buffers.
Making of TDP-43 constructs for eukaryotic expression
TDP-43∆1-50 was double digested with HindIII and SacII and cloned into pZsYellow C1 (Clontech, CA, USA) vector using Tardbp ZsY_Xho_318F and Tardbp ZsY_Sal_496R primers. The CDS of Tardbp variant 1 encoding the full length protein of TDP-43 (FL-TDP-43) of 43 kDa (414 AA) was sub cloned into pEGFP-N1 vector (Clontech, CA, USA) using Tardbp_ HindIII_318F forward and Tardbp_SacII_1575R reverse primers. FL-TDP-43 was cloned upstream of the C-terminal green fluorescent protein tag.
Transient transfection with sense TDP-43 (FL-TDP-43) and TDP-43∆1-50
GC-1 SPG cell line, a mouse spermatogonia-derived immortalized cell line that resembles type B spermatogonia and primary spermatocytes, was used in this study. A recent exhaustive microarray analysis of the gene expression of these cells confirmed them to be of spermatogonia origin.[22] Plasmid DNA with different TDP-43 constructs such as TDP-43∆1-50-pZsYellow1 C1 and FL-TDP-43-pEGFP-N1 plasmids were transfected into mammalian cells using Lipofectamine 2000 transfection reagent following manufacturer’s protocol. Transgene expression was tested 18–48 h post transfection. pEGFP-N1 and pZsYellow1 C1 plasmids were used as vector controls and TDP-43∆1-50-pZsYellow1 C1 and FL-TDP-43-pEGFP-N1 plasmids were used as tests for single and cotransfection experiments.
Protein extraction from GC1 cells
24 h post transfection, the growth medium was removed from the adherent mammalian cells (approximately 2 × 107 cells) and was rinsed thrice with sterile PBS containing KH2PO4, Na2HPO4, NaCl, KCl, Tween 20 followed by protein isolation by the addition of 1 ml of RIPA buffer (RIPA buffer (50 mM Tris-HCl [pH 8.0], 150 mM sodium chloride, 1% Igepal, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) having 10 mL protease inhibitor cocktail following standard protocol. For the extraction of nuclear and cytosolic protein fractions, 20 million transfected cells were harvested and washed thrice with sterile ice-cold PBS. The cell pellet was resuspended in a hypertonic buffer A (10 mM HEPES (pH 7.5), 10 mM KCl, 1.5 mM MgCl2, 0.5 M Dithiothreitol), containing a protease inhibitor cocktail. Cells were subjected to centrifugation at 1000 rpm for 5 min and the pellet was lysed in ice-cold buffer A, containing 0.5% NP-40 with protease inhibitor cocktail, on ice for 10 min. The nuclei were pelleted by centrifugation for 2 min at 3000 rpm at 4°C. Cytoplasmic protein in the supernatant was transferred into a fresh tube and stored at −20°C. The nuclear pellets were washed with buffer A without NP-40 and resuspended in buffer C (20 mM HEPES (pH 7.9), 420 mM NaCl, 0.2 mM EDTA, 1.5 mM MgCl2, 0.5 M Dithiothreitol, and 25% glycerol) containing protease inhibitor cocktail. Nuclei were incubated on ice for 30 min followed by periodic vortexing. The supernatant containing the nuclear protein was collected by spinning at 14,500 rpm at 4°C for 10 min and frozen immediately.
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE)
Protein extracted from cell lines was subjected to heat denaturation at 95°C for 5–15 min after a 1:2 dilution with Laemmli buffer. The denatured samples were centrifuged before loading into the wells and separated at constant voltage (100 V) on a 10–12% SDS-PAGE in a Biorad miniprotean gel electrophoresis tank containing Tris Glycine buffer.[23] The gels were stained with Coomassie Brilliant Blue R-250 or were used for immunoblotting.
Western blot analysis
The gels were electro-transferred to PVDF membranes following the Towbins method. The blot was blocked for 2 h in a blocking buffer containing 5% BSA/milk or 3% gelatin in phosphate buffered saline with 0.1% (v: v) Tween 20 (PBST). The membranes were washed in three changes of PBST and were incubated at room temperature for 2 h in the primary antibody (TARDBP rabbit polyclonal), diluted 1:1000 in PBST. After three washes in PBST, the blots were incubated with peroxidase-conjugated anti-rabbit IgG (1:2000 in PBST) secondary antibody for 1 h. The blots were washed 3 times in PBST and developed with DAB. Secondary antibody control experiments were also done as above, without primary antibody incubation, to rule out secondary antibody based non specificities. β-ACTIN was used as the loading control, for which goat polyclonal anti-b ACTIN (1:500 in PBST) was used as the primary antibody and peroxidase conjugated antigoat IgG (1:1000 in PBST) as the secondary antibody.
Phoretix 1D software (Phoretix International Newcastle upon Tyne, UK) was used to quantify protein expression by densitometric analysis of the bands from three technical replicates each of the three biological replicates. Corresponding β-actin levels were used for normalization. Data presented indicate mean ± standard deviation of β-actin normalized values of band intensities and are represented as histograms. Student’s t-test was carried out to confirm significance of differences between the groups. P < 0.05 was considered as statistically significant.
Immunofluorescence to detect transgene expression
GC-1 cells were allowed to grow on sterile coverslips in 12-well culture dishes. 18 h post-transfection, GFP or ZsYellow-expressing, single- or co-transfected cells were fixed with 4% paraformaldehyde for 15 min. The fixed cells were rinsed thrice with PBS and subjected to permeabilization in 0.1% Triton ×−100 solutions for 10 min at room temperature. For evaluating the intrinsic expression of TDP-43, the untransfected cells were grown on coverslips, fixed and permeabilized as described above and were treated with blocker (1% BSA) and incubated in primary antibody (anti-TARDBP antibody at a dilution of 1 in 1000 in PBST) for 2 h at 4°C. The cells were then rinsed thrice with PBS to remove any non-specific primary antibody binding and were incubated in Alexa-488 or FITC-conjugated secondary antibody (1:200 dilution in blocker) for 1 h at room temperature. The cells incubated in secondary antibodies alone served as the negative control. The cells were washed with PBS and nuclei were stained with 0.25 mg/mL PI stain for 5 min or Hoechst 33342 (10 mg/mL) for 5–15 min with incubation in dark. The excess stain was removed by PBS washing. The coverslip containing the treated cells was mounted on a clean glass slide containing 50% glycerol: saline solution for imaging using a confocal microscope (Leica TCS-SP2 AOBS system, Germany).
Confocal imaging
A confocal microscope was used to image the transgene expression in single and co-transfected cells. Excitation sources on the SP2 AOBS system include HeNe (633 nm/10 mW), red diode (561/20 mW), and Ar (458/5 mW, 476/5 mW, 488/20 mW, and 514/20 mW) lasers. For Fluorescence resonance energy transfer (FRET) analysis in cotransfected cells, bleed through caused due to overlapping spectra of the donor/acceptor pair EGFP/ZsYellow was minimized by modifying the slit bandwidths of the Leica SP2 instrument. The modifications adopted for imaging are as follows-laser excitation of 488 nm for EGFP, 514 nm for ZsYellow1 and 543 nm for PI; emission was collected at 520 nm, 525–560 nm and 615 nm for EGFP, ZsYellow1 and PI, respectively. Sequential acquisition of EGFP and ZsYellow1 fluorescence was obtained and superimposed, to depict co-localization.
Protein threading and molecular docking
To predict the interaction between TDP-43∆1-50 and FL-TDP-43, protein folding or threading was performed with MUlti-Sources ThreadER (MUSTER) algorithm (http:// zhang.bioinformatics.ku.edu/MUSTER). This algorithm provided the primary structure of the target peptide with the secondary structures of highly similar template sequences in the Protein database (PDB). The alignment requiring minimum free energy between the target and the template sequence was returned and a 3D model of the interacting protein complex was generated based on the Glide score (G score) of 45.
RESULTS
Directional cloning, expression and purification of TDP- 43∆3-183
A schematic diagram of TDP-43 highlighting various functionally important domains is presented in [Figure 1]. NRRM1 domain starts from the 3rd amino acid to the 183rd AA and spans the N terminus and the RRM1. Using Tardbp 337F forward and Tardbp 878R reverse primers [Table S1] a 540 bp fragment that codes for the NRRM1 (N-terminus and RRM1) domain of 180 AA (TDP-43∆3-183) was amplified from mouse testicular cDNA. This fragment was cloned into pET 100/D prokaryotic expression vector. The 21 kDa recombinant protein (TDP-43∆3-183) was expressed downstream the ×6 His tag as a 27 kDa fusion protein in the soluble and insoluble protein fractions of IPTG-induced culture, (Uninduced culture served as the negative control). From the second lysate of insoluble proteins in induced culture in the 200- and 300 mM imidazole fractions of buffer containing 8M urea, TDP-43∆3-183 was purified. These purified fractions in eluate were desalted by dialysis and concentrated using PD10 columns. A western blot using an anti-TARDBP antibody confirmed the purified protein of 27 kDa to be TDP-43∆3-183 [Figure S1a].
Does TDP-43∆3-183 domain dimerize in vitro?
We evaluated whether the NRRM1 domain (TDP-43∆3-183) alone can mediate dimerization of TDP-43 in vitro. The desalted and concentrated purified peptide, TDP-43∆3-183, was denatured and subjected to electrophoresis on an SDS-PAGE gel, under reducing and non- reducing conditions. Prior to electrophoresis, one fraction of the heat denatured protein was treated with β-mercaptoethanol to break disulfide bonds thereby removing any secondary structures. The second fraction was the non-reduced control. The treated and untreated fractions were electrophoresed and stained using Coomassie Blue R-250. Both reduced and non-reduced fractions displayed the 27 kDa protein and no higher sized dimers or complexes were detected in either condition, indicating that TDP-43∆3-183 domain did not self-dimerize [Figure 2a].

- A schematic representation of TDP-43 protein with the location of its domains. ☒r localization Signal, RRM1 and RRM2: RRM1 and RRM2, NES: Nuclear export signal).
Directional cloning, expression and purification of recombinant TDP-43∆1-50
150 bp fragment of TDP-43 (TDP-43∆1-50) that encodes the N-terminal region lacking the NLS peptide was directionally cloned into pET 102/D expression vector. TDP-43∆1-50 of 5 kDa was expressed as a recombinant fusion protein of 18 kDa, downstream the His-Patch Thioredoxin tag of 13 kDa, was eluted from the IPTG-induced bacterial culture of soluble protein fractions using buffers containing 100- and 200 mM imidazole [Figure S1b]. The commercially available TARDBP antibody raised against the antigenic peptide corresponding to the first 50 AA of TDP-43. This N terminal peptide, (TDP-43∆1-50), was used as a blocking peptide to the antibody.
In vitro dimerization of TDP-43∆1-50 with TDP-43 isoforms in GC-1 protein lysates
GC-1 cells maintained in our laboratory were recently subjected to microarray analysis to confirm that they are indeed of spermatogonia origin.[22] Further, we also confirmed the expression of ddx4 and gfrα-1 in these cells to confirm their germ cell origin [Figure S2] RT-PCR analysis of Protein lysates of GC-1 cells were subjected to electro-blotting onto PVDF membranes. Before treating the membrane with anti-TARDBP-1 antibody, the antibody was incubated with the recombinant TDP-43∆1-50 peptide (1.25 mg/mL antibody: 5.86 mg/100 mL peptide) for 30 min. The primary antibody, which was not pre-incubated with the peptide served as the control of the experiment . β-Actin was used as the loading control. Secondary antibody control, wherein the membrane was treated with secondary antibody alone, was performed as a negative control. The intensity of the band corresponding to the 43 kDa isoform of TDP-43 enhanced upon incubation with the peptide and this observation inferred that TDP-43 dimerizes with the N-terminal peptide, TDP-43∆1-50 [Figure 2b].
TDP-43∆1-50 dimerizes with FL-TDP-43 in GC-1 cells
Both in the untransfected, as well as transfected GC1 cells, endogenous TDP-43 isoforms, were detected in 43 kDa (isoform 1:NP_663531.1 34) and 34 kDa (Isoform 2-6 as: NP_001008545.1, NP_001003899.1, NP_001008546.1, NP_001003898.1, and NP_001292354.1). A 70 kDa band [Figure 2c] in the lysates of cells transfected with sense constructs of TDP-43 (FL-TDP-43-GFPN1) refers to the expression of recombinant full length TDP-43 upstream of the GFP tag (27kDa). On the other hand, the cells were transfected with TDP-43∆1-50-ZsYellow constructs, and the fusion protein of 33 kDa was detected in the cytoplasm [Figure 2d, lane 4]. TDP-43∆1-50, which lacked the NLS sequence retained the peptide within the cytosol and a very small amount was detected in the nucleus [Figure 2d, lanes 3 and 4]. As expected, the endogenous TDP-43 (43 kDa isoform) was detected in the nuclear fractions with negligible expression in the cytosolic lysates [Figure 2d]. Although endogenous TDP-43 is diffusely distributed within the nuclei of untransfected GC-1 cells, cells transfected with the empty vectors, pEGFPN1 or pZsYellow1 C1 expressed the reporter protein in the nuclei as well as in the cytoplasm. Furthermore, FL-TDP-43 transfected cells exhibited distinct nuclear localization of the protein [Figure 3a-e]. When cotransfected, both FL-TDP-43 and TDP-43∆1-50 were together in the nuclei. Interestingly, though the NLS, spanning AA 80–98 in FL-TDP-43 was absent in TDP-43∆1-50, the full length protein dimerized with the N terminal peptide and translocated TDP-43∆1-50 to the nucleus [Figure 3g and f] represents double transfection with the empty vectors.

- (a) SDS-PAGE gel showing TDP-43∆3-183 of 27 kDa detected under reduced (r) and non-reduced (NR) conditions. (b) Western blot with anti TARDBP antibody showing enhanced intensity of 43 kDa (<<) isoform of TDP-43 in GC-1 protein lysates after pre-incubation of the antibody with recombinant TDP-43∆1-50 peptide. 1 - Secondary antibody control, 2 - β-ACTIN, 3 - Antibody alone, 4 - antibody: peptide in 1:100 dilution, M- protein marker. (c and d)-Western blots with protein lysates of untransfected and transfected GC-1 cells using anti-TARDBP antibody. Endogenous TDP-43 isoforms (<<) and GFP-tagged FL-TDP-43 of 70 kDa (•) in (c) and TDP-43∆1-50 –pZsYellow fusion protein of 33 kDa (*) in (d) are shown in the top panels. β-ACTIN of 42 kDa is presented at the bottom.

- Immunofluorescence showing TDP-43 localization in (a) untransfected, (β-e) single-transfected, and (f and g) cotransfected GC-1 cells. Note the nuclear localization of TDP-43 in (c) and predominant cytosolic protein in (e), compared to empty vector control (b and d). Yellow fluorescence is observed in the nuclei of cells colocalizing TDP-43∆1-50 and FL-TDP-43. ZsY denotes pZsYellow.
FRET analysis to show TDP-43∆1-50 and FL-TDP-43 interactions
To confirm the interaction between TDP-43∆1-50 and FL-TDP-43 in GC-1 cells co-expressing these proteins, FRET analysis of fluorochrome tagged TDP-43 protein was carried out. The donor and acceptor in the FRET pairs were GFP tagged FL-TDP-43 and pZsYellow tagged TDP-43∆1-50, respectively. [Figure 4a] represents the negative control experiment for FRET analysis. Three different images of single-transfected cells expressing GFP (GFP or FL-TDP-43-GFP) were captured with donor excitation-donor emission, donor excitation-acceptor emission and acceptor excitation-acceptor emission wavelengths. Fluorescence observed at the donor excitation-donor emission wavelength of 488 nm/520 nm was not detected with donor excitation-acceptor emission (488/525–560 nm) and acceptor excitation-acceptor emission (514/525–560 nm) wavelengths. The absence of yellow fluorescence in cells expressing eGFP confirms the absence of bleed through. FRET between FLTDP-43-GFP and TDP-43∆1-50-ZsYellow in cotransfected cells excited at 488 nm was clearly visible at 488/520–560 nm [Figure 4b]. The efficiency of the FRET was calculated by measuring the fluorescence intensity in protein lysates using a Perkin Elmer Luminescence Spectrometer LS50B. Lysates of GFP and FL-TDP-43-GFP expressing cells were excited at the donor excitation wavelength of 488 nm and emission intensity was measured at the acceptor emission peak of 539 nm. Increased fluorescence intensity was observed at 539 nm in samples containing the FRET pairs GFP/ZsYellow or FL-TDP-43-GFP/TDP-43∆1-50-ZsYellow. The increased intensity at the acceptor emission peak is due to FRET from the donor to the acceptor. The histogram representing the donor intensities at 539 nm and shows a FRET efficiency of 0.1 [Figure 4c].

- Confocal images for fluorescence resonance energy transfer studies in transfected GC-1 cells. (a) is the negative control experiment showing absence of fluorescence at the donor excitation/acceptor emission of 488 nm/525–560 nm (GFP/ZsY) and acceptor excitation/ acceptor emission of 514 nm/525–560 nm (ZsY/ZsY) in single-transfected, GFP-expressing cells namely, FL-TDP-43-GFP and GFP. (b) shows fluorescence resonance energy transfer between FL-TDP-43-GFP (donor) and TDP-43∆1-50-ZsYellow (acceptor) in the tile marked as GFP/ZsY. (c) is a histogram depicting the intensity of acceptor emission at 539 nm upon excitation at 488 nm. eGFP and FL-TDP are the donors and ZsY and N`-ZsY are the acceptors. TDP-43∆1-50 is denoted as N`-ZsY. (d) In silico model of predicted homodimerization of TDP-43∆1-50 (pink spheres) with FL-TDP-43 (green). The amino acids on each chain within a distance of 2A° are enlisted. The occurrence of amino acid residue in both the chains implies its role in dimerization.
Protein threading and molecular docking studies on TDP-43∆1-50 and FL-TDP-43 interaction
To predict the interaction between TDP-43∆1-50 and FL-TDP-43, protein folding or threading was performed with the MUSTER algorithm. This algorithm provided the primary structure of the target peptide with the secondary structures of highly similar template sequences in the PDB. The alignment requiring minimum free energy between the target and the template sequence was returned and a 3D model of the interacting protein complex was generated based on the Glide score (G score) of 45. AA residues of the target and template protein that were found at the interacting interface within a distance of 2A° are enlisted in [Figure 4d]. Using these in silico predictions, the sixth AA, Arginine (R6) was found to be the common residue at the interacting interface of TDP-43∆1-50 and FL-TDP-43 and hence crucial for homodimerization.
DISCUSSION
The functional role of TDP-43 in facilitating several cellular processes and pathological fibrillar formation in diseased conditions are ascribed to the homo-dimerization abilities of TDP-43. Though the disease biology of TDP-43 was studied widely, the major focus has been on the glycine-rich C-terminal that harbors all the ALS-associated TDP-43 mutations[24-27] and the Q/N-rich domains that promote TDP-43 aggregation.[28,29] It is reported that the CTD of TDP-43 behaved like a prion and could promote aggregation and mislocalization and that mutations in this region led to abnormal protein-protein interactions.[30,31] However, HEK cells transfected with a 25kDa recombinant TDP-43 region similar to the caspase-cleaved truncated TDP-43 C-terminal product generated toxic cytoplasmic aggregations within the cells possibly through a toxic gain of function. But this C-terminal cytoplasmic aggregation occurred independently and it neither showed any biological activity nor did it inhibit the function of full length TDP-43 by aggregation formation.[32] However, our co-transfection experiments indicated that the N terminal peptide (TDP-43∆1-50) and FL-TDP-43 dimerized in GC1 cells, which resulted in the translocation of NLS negative TDP-43∆1-50 to the nucleus which indicates that the homodimerization domain of TDP-43 lies within AA 1–50, whereas the nuclear localization domain appears to be located posterior to this region. Contrary to the self-aggregation of CTD without affecting the biological function of full length TDP-43, the N-terminal peptide engaged in direct interaction with full length TDP-43. Furthermore, the 27 kDa NRRM1 domain of TDP-43 also failed to self-dimerize in vitro.
Shiina et al. in 2010[33] had shown the necessity of the NRRM1 domain for homo-dimerization. Studies by Buratti and Baralle[7] also demonstrated that the deletion of NRRM1 halted the capability of TDP-43 to bind to RNA. They also revealed that the RRM1 sequence spanning residues 106–175 in TDP-43 is ineluctable for RNA binding. Another study employing single-molecule fluorescence uncovered that TDP-43 NTD was well-folded, thermodynamically stable and underwent reversible oligomerization, and interaction of folded NTD with full length TDP-43 boosted the propensity of inherently unfolded CTD to form pathological aggregation.[34] Our study using the NRRM1 construct showed the absence of higher molecular weight aggregates on SDS-PAGE of a heat-denatured and mercaptoethanol-reduced fraction of recombinant NRRM1 pointed out the inability of NRRM1 to mediate self-dimerization. In addition, the normal function of NLS was also found to be disturbed by caspase cleavage at Asp 89, which causes the accumulation of mislocalized TDP-43 in the cytoplasm.[8] Knocking down of karyopherin-b1 and cellular apoptosis susceptibility protein (factors that regulate NLS) by siRNA mediated silencing in Human neuroblastoma SHSY-5Y also caused significant cytoplasmic accumulation of TDP-43.[35]
CFTR exon 9 splicing assay by Zhang’s group[32] revealed that the deletion of the first 75 AA residues of TDP-43 considerably weakened its major biological activities such as homodimerization, splicing, as well as self-interaction, of which the first 10 AA are critical in full length TDP-43 aggregation in both normal and pathological conditions.[36] In vitro and in silico studies conducted by the same group also revealed two major functions attributed to the N terminus of TDP-43; protein interaction essential for its normal biological activity and TDP-43 inclusion formation in pathological conditions, which in turn, impair neuronal growth. They had also shown in the mouse neuronal cells the pernicious role of residues such as Arginine 6, Valine 7, Threonine 8 and Glutamate 9 involved in protein folding and homodimerization of TDP-43. In agreement with this, our in silico prediction study also showed that the 6th Arginine residue positioned at the interface of the interacting proteins, FL-TDP-43 and TDP-43∆1-50, was critical for dimerization. Furthermore, size exclusion chromatography (SEC) analysis on several mutants of TDP-43 based on the monomeric structure revealed that the residues of Leu71-Val74 in the b7-strand are mostly clustered in the dimeric interface and are critical for TDP-43 NTD dimerization.[37] However, the polymerization property of NTD can be interrupted by the phosphomimetic substitution at S48, as a consequence the RNA splicing property of the molecule is hampered.[38]
Although an NLS region is absent in TDP-43∆1-50, it co-localized with FL-TDP-43 within the nuclei of co-transfected cells. This confirms that dimerization with FL-TDP-43 is ample rather than the presence of an NLS sequence for the nuclear translocation of cytosolic TDP-43∆1-50. Remarkably, our FRET analysis and in silico predictions, along with dimerization of TDP-43∆1-50 with TDP-43 isoforms confirmed the interaction between TDP-43∆1-50 and FL-TDP-43 in mammalian spermatogonial cells. Furthermore, the NRRM1 domain, that was reported to mediate TDP-43 dimerization, did not self-dimerize. However, in agreement with the observation of Zhang,[36] the shorter N terminal peptide of TDP-43, which was NLS negative, was sufficient for homodimerization. Since the oligomerization of TDP-43 is regulated by the NTD in a concentration-dependent manner which controlled its nucleic acid binding properties,[39] the physiological implications of the N-terminal peptides of TDP-43 oligomerization with full-length TDP-43 need to be investigated.
CONCLUSION
A deletion construct of TDP-43 lacking the first two amino acids (TDP-43∆3-183) was not capable of homodimerization, while TDP-43∆1-50 dimerized with full length TDP-43 and translocated to the nucleus. This indicates the importance of amino acids 1-50 of TDP-43 in dimerization and invokes the physiological and pathological implications of the truncated versions of TDP-43 dimerizing with full-length TDP-43 and gaining access to the nucleus.
Declaration of patient consent
Patient’s consent not required as there are no patients in this study.
Conflicts of interest
There are no conflicts of interest.
SUPPLEMENTARY FIGURES

- Western blot showing (a) purified fusion protein TDP-43∆3-183 in lane 2. Lane 1 is left blank. (b) Purified TDP-43∆1-50 fusion protein of 18 kDa eluted in buffer containing imidazole. 1 – flow through, 2-wash, 3–7 - 100 mM imidazole fractions, 8 and 9 - 200 mM fractions. Lanes marked M represents the protein marker.
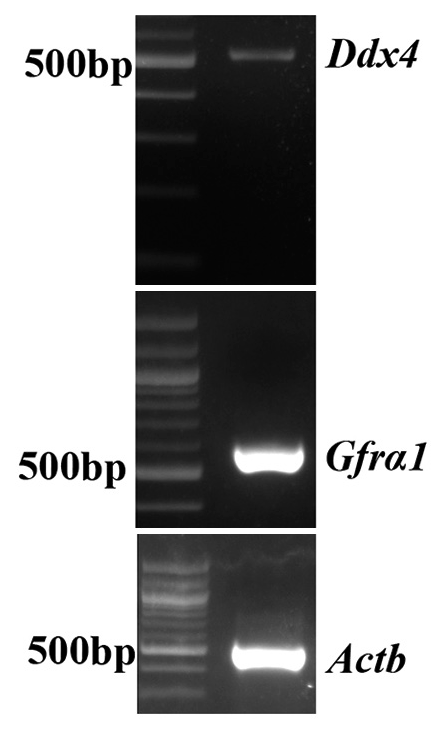
- Polymerase chain reaction showing the amplification of Gfrα-1, Ddx4, and β-Actin from GC-1 cells.
| Primer name | Primer sequence (5'-3') |
|---|---|
| Tardbp_318 F | ATTTAAGCAAAGATGTCTGAA |
| Tardbp_1575 R | CCTACATTCCCCAGCCAGA |
| Tardbp_2186 R | TTACAGCACTACTTTCAATGA |
| Tardbp_2268 R | CACAGGCTGCGTCTTTGA |
| Tardbp_641 F | TCTCCCCTGGAAAACAACTG |
| Tardbp_pET_exp_496R | GTCGGACTCCTCTCATTCA |
| Tardbp_ZsY_XhoI_318F | GAACGTCTCGAGATTTAGCAAAGATGTCTGAA |
| Tardbp_ZsY_SalI_496R | TGAATATGTCGACTTAACACTGAGACACGGGAT |
| Tardbp_GFP_HindIII_318F | GCCCGCAAGCTTATTTTAGCAAAGATGTCTGAA |
| Tardbp_GFP_SacII_1575R | TATAATCCGCGGCTTTCCCCAGCCAGAAGACTT |
| Tardbp_AS_FL_HindIII_1575F | GCCCGCAAGCTTCTACATTCCCCAGCCAGA |
| Tardbp_AS_FL_SacII_318R | TATAATCCGCGGGCAAAGATGTCTGAATATATTCGGGTA |
| Tardbp_AS_3utr_Hind_2261F | GCCCGAAAGCTTCACGGCGCAGCCTGTGCAGCGTGAT |
| β Actin F | TGTGATGGTGGGAATGGGTCAG |
| β Actin R | TTTGATGTCGCGCACGATTTCC |
| Dd×4-F | GAGGATGAGGACTCCATCTTTG |
| Dd×4-R | CTCCCTGGAGTAGCACACAATA |
| Gfrα-1 F | TCTGTCCCCTGTCCTCTTGTAT |
| Gfrα-1 R | GACCACACCCACTCTCCTCTAC |
Financial support and sponsorship
Intramural Research Grants from Rajiv Gandhi Centre for Biotechnology.
References
- Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus Type 1 TAR DNA sequence motifs. J Virol. 1995;69:3584-96.
- [CrossRef] [PubMed] [Google Scholar]
- TDP-43 is a developmentally regulated protein essential for early embryonic development. J Biol Chem. 2010;285:6826-34.
- [CrossRef] [PubMed] [Google Scholar]
- Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J Neurosci. 2007;27:10530-4.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat Struct Mol Biol. 2013;20:1443-9.
- [CrossRef] [PubMed] [Google Scholar]
- Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001;20:1774-84.
- [CrossRef] [PubMed] [Google Scholar]
- TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: An important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem. 2005;280:37572-84.
- [CrossRef] [PubMed] [Google Scholar]
- Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem. 2001;276:36337-43.
- [CrossRef] [PubMed] [Google Scholar]
- TDP-43-induced death is associated with altered regulation of BIM and Bcl-xL and attenuated by caspase-mediated TDP-43 cleavage. J Biol Chem. 2011;286:13171-83.
- [CrossRef] [PubMed] [Google Scholar]
- Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem. 2008;283:13302-9.
- [CrossRef] [PubMed] [Google Scholar]
- TDP-43 condensation properties specify its RNA-binding and regulatory repertoire. Cell. 2021;184:4680-96.e22.
- [CrossRef] [PubMed] [Google Scholar]
- TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011;30:277-88.
- [CrossRef] [PubMed] [Google Scholar]
- Loss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurones. Neuropathol Appl Neurobiol. 2014;40:670-85.
- [CrossRef] [PubMed] [Google Scholar]
- Biomarkers for diseases with TDP-43 pathology. Mol Cell Neurosci. 2019;97:43-59.
- [CrossRef] [PubMed] [Google Scholar]
- Updated TDP-43 in Alzheimer's disease staging scheme. Acta Neuropathol. 2016;131:571-85.
- [CrossRef] [PubMed] [Google Scholar]
- Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD) Acta Neuropathol. 2014;127:423-39.
- [CrossRef] [Google Scholar]
- Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol. 2013;74:20-38.
- [CrossRef] [PubMed] [Google Scholar]
- Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain. 2019;142:1503-27.
- [CrossRef] [PubMed] [Google Scholar]
- Immunolocalization of TAR DNA-binding protein of 43 kDa (TDP-43) in mouse seminiferous epithelium. Mol Reprod Dev. 2017;84:675-85.
- [CrossRef] [PubMed] [Google Scholar]
- Cis-requirement for the maintenance of round spermatid-specific transcription. Dev Biol. 2006;295:781-90.
- [CrossRef] [PubMed] [Google Scholar]
- Aberrant expression of TAR DNA binding protein-43 is associated with spermatogenic disorders in men. Reprod Fertil Dev. 2016;28:713-22.
- [CrossRef] [PubMed] [Google Scholar]
- Loss of TDP-43 in male germ cells causes meiotic failure and impairs fertility in mice. J Biol Chem. 2021;297:101231.
- [CrossRef] [PubMed] [Google Scholar]
- Revisiting the characteristics of testicular germ cell lines GC-1(spg) and GC-2(spd)ts. Mol Biotechnol. 2021;63:941-52.
- [CrossRef] [PubMed] [Google Scholar]
- Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680-5.
- [CrossRef] [PubMed] [Google Scholar]
- TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63:535-8.
- [CrossRef] [PubMed] [Google Scholar]
- TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668-72.
- [CrossRef] [PubMed] [Google Scholar]
- TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol. 2008;63:538-42.
- [CrossRef] [PubMed] [Google Scholar]
- Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum Mol Genet. 2009;18:3353-64.
- [CrossRef] [PubMed] [Google Scholar]
- Interaction with polyglutamine aggregates reveals a Q/N-rich domain in TDP-43. J Biol Chem. 2010;285:26304-14.
- [CrossRef] [PubMed] [Google Scholar]
- Cellular model of TAR DNA-binding protein 43 (TDP-43) aggregation based on its C-terminal Gln/Asn-rich region. J Biol Chem. 2012;287:7512-25.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front Mol Neurosci. 2019;12:25.
- [CrossRef] [PubMed] [Google Scholar]
- Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci U S A. 2009;106:7607-12.
- [CrossRef] [PubMed] [Google Scholar]
- TDP-43 dimerizes in human cells in culture. Cell Mol Neurobiol. 2010;30:641-52.
- [CrossRef] [PubMed] [Google Scholar]
- The N-terminal domain of ALS-linked TDP-43 assembles without misfolding. Angew Chem Int Ed Engl. 2017;56:12590-3.
- [CrossRef] [PubMed] [Google Scholar]
- Nuclear import impairment causes cytoplasmic trans-activation response DNA-binding protein accumulation and is associated with frontotemporal lobar degeneration. Brain. 2010;133:1763-71.
- [CrossRef] [PubMed] [Google Scholar]
- The dual functions of the extreme N-terminus of TDP-43 in regulating its biological activity and inclusion formation. Hum Mol Genet. 2013;22:3112-22.
- [CrossRef] [PubMed] [Google Scholar]
- The N-terminal dimerization is required for TDP-43 splicing activity. Sci Rep. 2017;7:6196.
- [CrossRef] [PubMed] [Google Scholar]
- A single N-terminal phosphomimic disrupts TDP-43 polymerization, phase separation, and RNA splicing. EMBO J. 2018;37:e97452.
- [CrossRef] [Google Scholar]
- The N-terminus of TDP-43 promotes its oligomerization and enhances DNA binding affinity. Biochem Biophys Res Commun. 2012;425:219-24.
- [CrossRef] [PubMed] [Google Scholar]




